

估计阅读时长: 15 分钟进行生物化学代谢反应网络的模拟计算,可以分为三种技术路线:基于线性规划做优化的FBA方法,基于常微分方程组求解的动力学模拟方法,以及最近发展的基于图神经网络做模拟计算的深度学习计算方法。在下面的表格中,在这里进行比较和总结了上面所提到的三种计算分析方法各自的计算原理和应用领域: 计算方法 原理 优势 适用场景 通量平衡分析(FBA) 基于约束条件(如化学计量矩阵、酶容量限制)和线性规划,在假设代谢网络处于稳态(即代谢物浓度不变)的前提下,计算代谢通量的分布,通常以最大化特定目标(如生物量生长)进行优化 1. 无需详细的酶动力学参数,特别适合大规模网络研究。2. 计算速度快,可系统性地预测基因敲除或环境扰动下的表型变化。3. 广泛应用于指导代谢工程,优化目标产物合成。 追求快速评估和全局优化:如果你的研究目标是在基因组尺度上快速评估微生物在不同条件下的生长或产物合成潜力,并且难以获取详细的动力学参数,FBA是一个非常实用的起点 动力学模拟 基于质量作用定律等构建常微分方程组(ODEs),描述每个代谢物浓度随时间变化的动力学过程,通过数值方法求解方程组 1. 能够捕捉代谢物浓度和通量的瞬态动态变化,揭示更精细的调控机制2. […]
估计阅读时长: 7 分钟https://github.com/xieguigang/sciBASIC 在分布式哈希表网络之中,Peer节点之间进行分布式数据传输都是使用的B编码。B编码格式与JSON编码格式较为相似,均以“键:值”形式存储,我们可以将B编码的字符串整个内容理解为一个经过特殊编码的字典,或者一个近似的JSON。B编码与JSON编码,这两种编码都仅包含有4种最基础的数据类型:字符串类型,数值类型,数组类型与对象字典类型。 Order by Date Name Attachments DHT-dark-all • 416 kB • 638 click 2021年6月4日bdecode • […]
估计阅读时长: 10 分钟https://github.com/xieguigang/sciBASIC 根据积分表达式,微分方程的数值解关键在于微分方程的初值及计算微分方程式在tm(上一时刻)与tm+d(下一时刻)与坐标轴围成面积,若这个面积计算得越准确则得到的数值解也就越精确。微分表达式中与坐标轴围成的面积可表示如下,在实施算法的时候可以结合这个图更加直观点: 从上面的示意图可以看出,一段需要进行面积积分的曲线实际上是由多个梯形构成的多边形。那我们实际上只需要将这些梯形的面积都求出来,然后加起来就好了。 这里的梯形分割就是一种欧拉逼近的思想,欧拉逼近的几何意义,就是我们可以使用一段折线来近似的逼近一条曲线。 利用欧拉逼近,我们可以将一个精确的微分方程曲线 近似的使用线段来表示 Order by Date Name Attachments ODE_Trapezoidal • 30 kB • […]
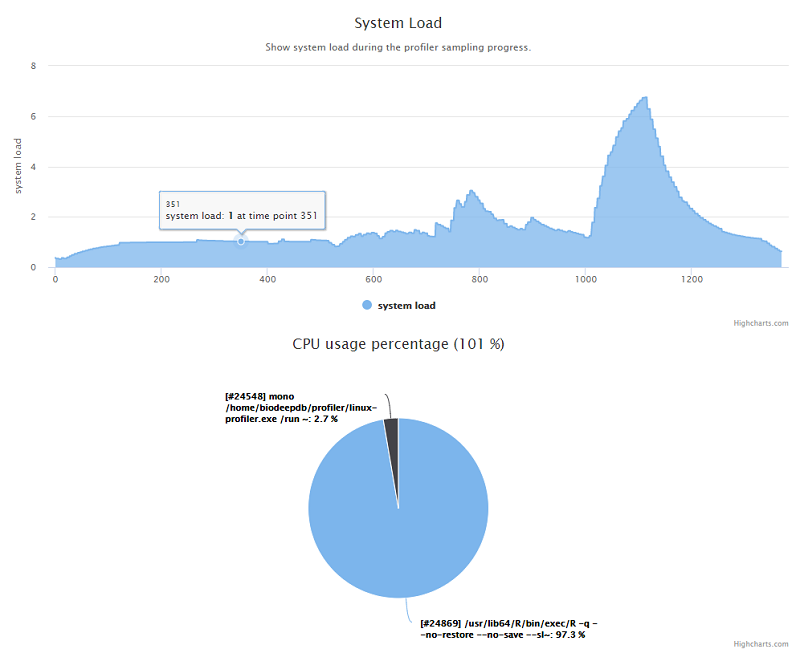
估计阅读时长: 6 分钟https://github.com/xieguigang/linux-profiler 废话不多说,首先给出一个 demo报告链接 给大家看看这个小工具的成品输出。 在去年的工作中,因为公司需要购买新的服务器做集群计算,需要一个工具来记录之前的服务器在数据分析上的性能瓶颈。于是花了两天的时间赶出来了这个专门应用于Linux系统的性能记录工具。这个小工具是一个开源项目,大家可以在Github上阅读这个开源项目(linux-profiler)的源代码。 Order by Date Name Attachments systemLoad • 53 kB • 751 […]
估计阅读时长: 5 分钟https://github.com/xieguigang/Darwinism 最近在做一个代谢组学的数据分析程序,由于需要被分析的质谱原始数据的计算量非常大,所以肯定会需要上并行计算。在并行计算中,分为两种模式:线程并行以及进程并行。 关于如果选择脚本代码的并行模式,我在这里借用了matlab文档网站里面的一张图来给大家做参考: 《Choose Between Thread-Based and Process-Based Environments》 Order by Date Name Attachments super_computing • […]

估计阅读时长: 2 分钟在BILIBILI上观看视频:《【GCModeller教程】KEGG代谢途径注释原理 (重置版)》 Order by Date Name Attachments kegg_annotation • 468 kB • 827 click 2021年5月30日release.mp4_20190921_225235.396 • […]

估计阅读时长: 2 分钟https://github.com/xieguigang/mzkit 在BILIBILI上观看视频:《【BioNovoGene Mzkit教程】代谢组学原始数据处理基础》 Order by Date Name Attachments profile_videocard • 211 kB • 754 click 2021年5月29日metabolims […]
估计阅读时长: 9 分钟前段时间由于工作的需要,会需要从一些网站上抓取数据用来做数据分析。在原来我进行网页爬虫开发的时候,一般会需要专门针对网页格式,使用大量的正则表达式进行内容的解析。由于你也知道,VisualBasic语言所开发的程序为一个编译好的Assembly文件,所以假若所需要爬取的网页格式变化了,我们就需要对代码做修改和重新编译。这个时候就会非常的不方便。 Order by Date Name Attachments ea5d2885-bba5-410f-b02b-0589613412ed • 12 kB • 752 click 2021年5月29日graphquery_Rscript • 36 […]

估计阅读时长: < 1 分钟https://github.com/rsharp-lang/R-sharp R#语言最开始的开发需求来自于对GCModeller的组件的调用需求。因为最开始GCModeller使用的是命令行模式进行运行,但是因为VB.NET语言为编译型语言,所开发的应用程序在发布之后,用户无法轻易的修改。自己对于一些比较个性化的数据分析,在引入R#语言之前,需要专门编写一段命令行代码跑GCModeller,会十分的不方便。所以后面就有了R#脚本语言的开发。 R#语言类似于R或者Matlab语言,也是一种向量化的编程脚本语言。其语法源自于R语言,同时也结合了一些TypeScript的语法,例如TypeScript之中的字符串插值语法就被引入了R#语言之中。 const words = ["world", "R# language", "GCModeller User"]; const hello = `hello ${words}!`; […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?