
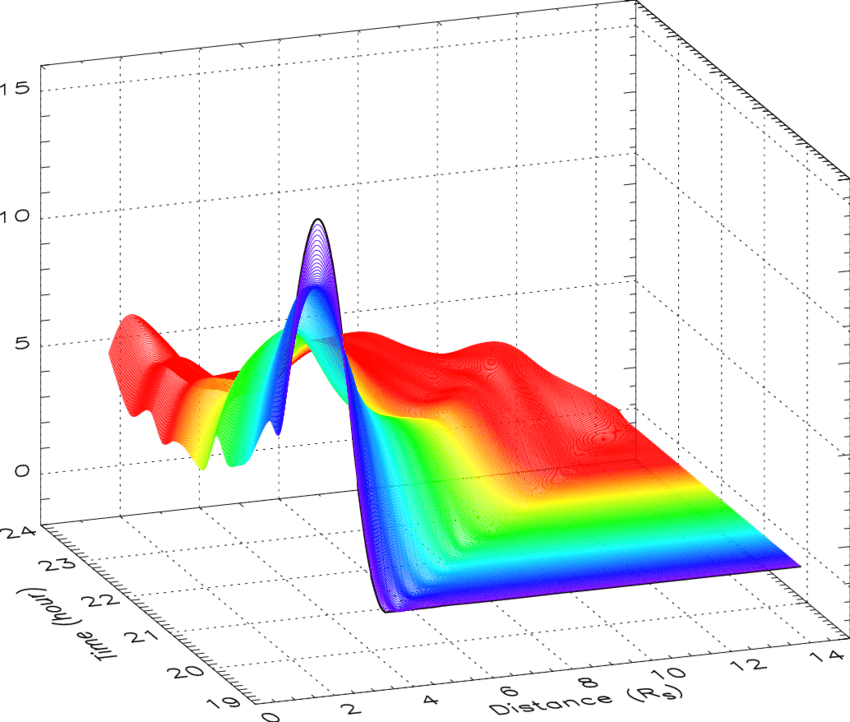
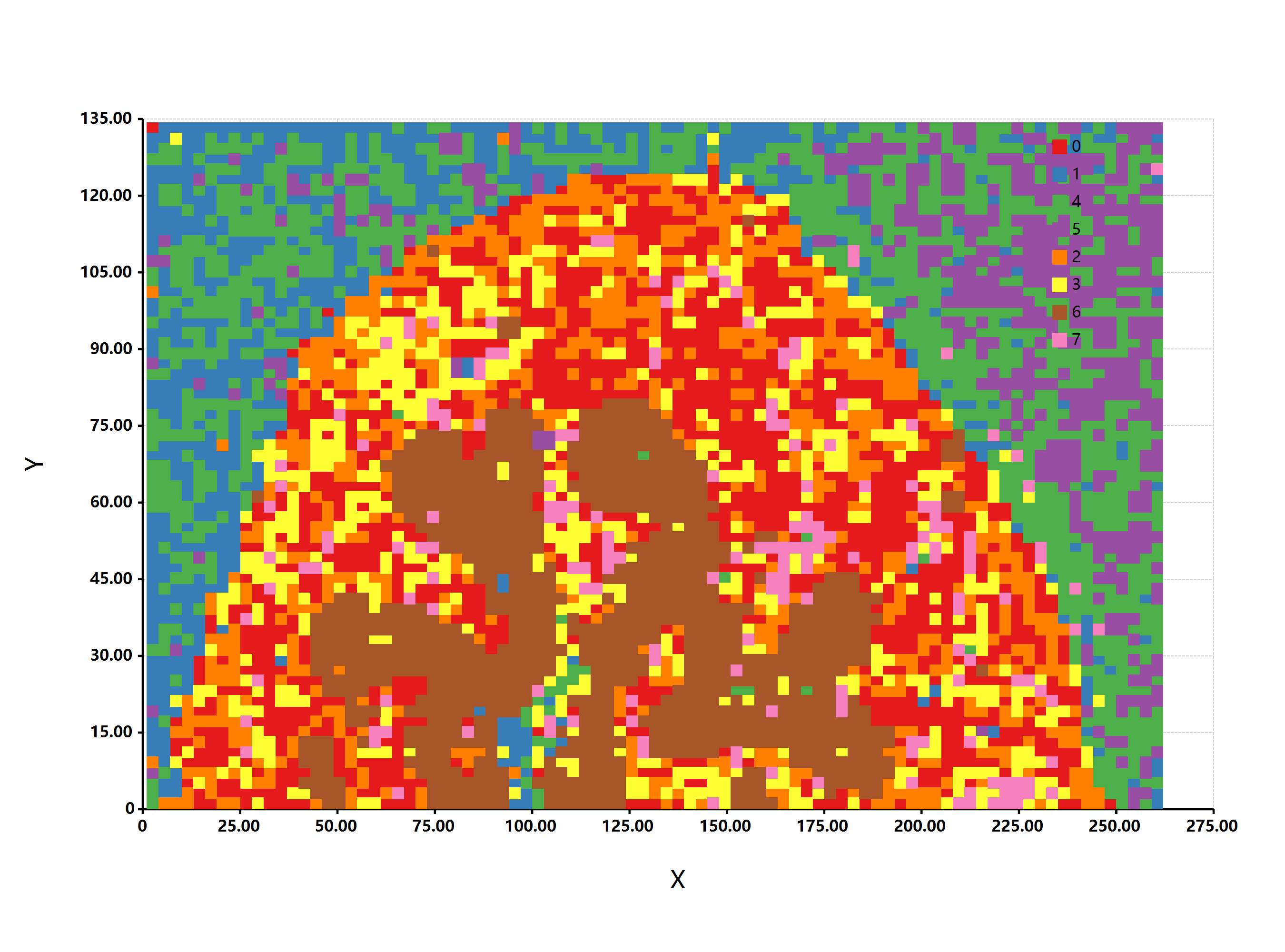
估计阅读时长: 7 分钟热图(Heat Map)是在二维空间中以颜色的形式显示一个现象的绝对量一种数据可视化技术。颜色的变化可能是通过色调或强度,给读者提供明显的视觉提示,说明现象是如何在空间上聚集或变化的。热图有两种完全不同的类别:聚集热图和空间热图。 在聚集热图中,幅度被排列成一个固定单元格大小的矩阵,其行和列是离散的现象和类别,行和列的排序是有意的,而且有些随意,目的是暗示聚集或描绘出通过统计分析发现的聚集。单元格的大小是任意的,但足够大,可以清晰可见。 相比之下,空间热图中某一量级的位置是由该量级在该空间中的位置所决定的,没有单元的概念,现象被认为是连续变化的。 Order by Date Name Attachments 2D-cubic-spline-interpolation-of-mass-profiles-from-1939-to-2354-UT-and-between-16 • 112 kB • 851 click […]
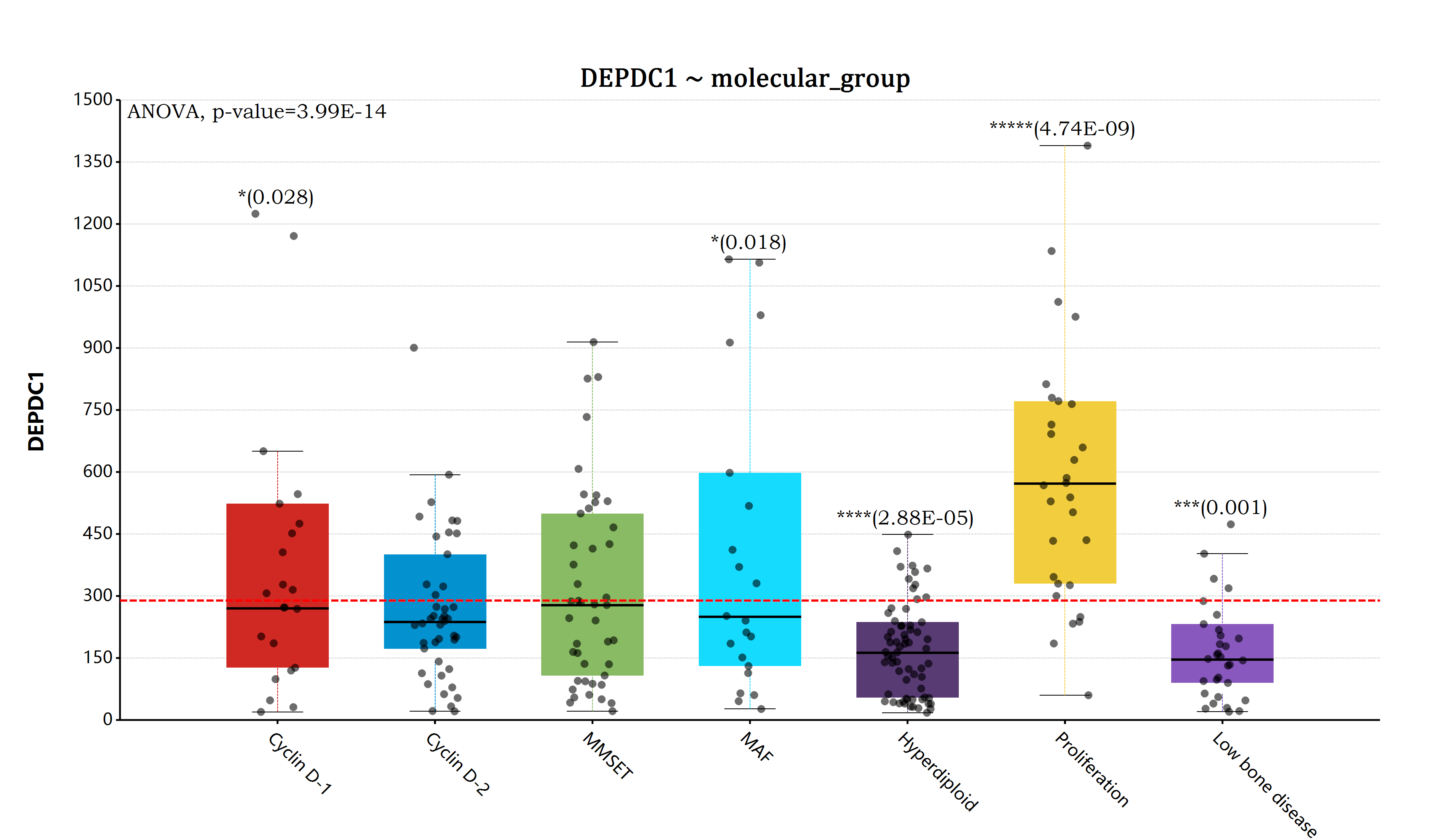
估计阅读时长: 14 分钟https://github.com/rsharp-lang/ggplot 在完成了前面所提到的ANOVA检验模块的代码开发编写工作之后,之前一直悬在我心里面的完善R#语言的ggplot统计作图功能的愿望现在终于实现了。在R#语言之中通过使用ggplot代码库进行相应的数据统计分析作图,目前已经变得和R语言之中的ggplot2程序包那样同样的简单和漂亮。 Order by Date Name Attachments myeloma_bar • 196 kB • 772 click 2022年5月29日myeloma_box • […]
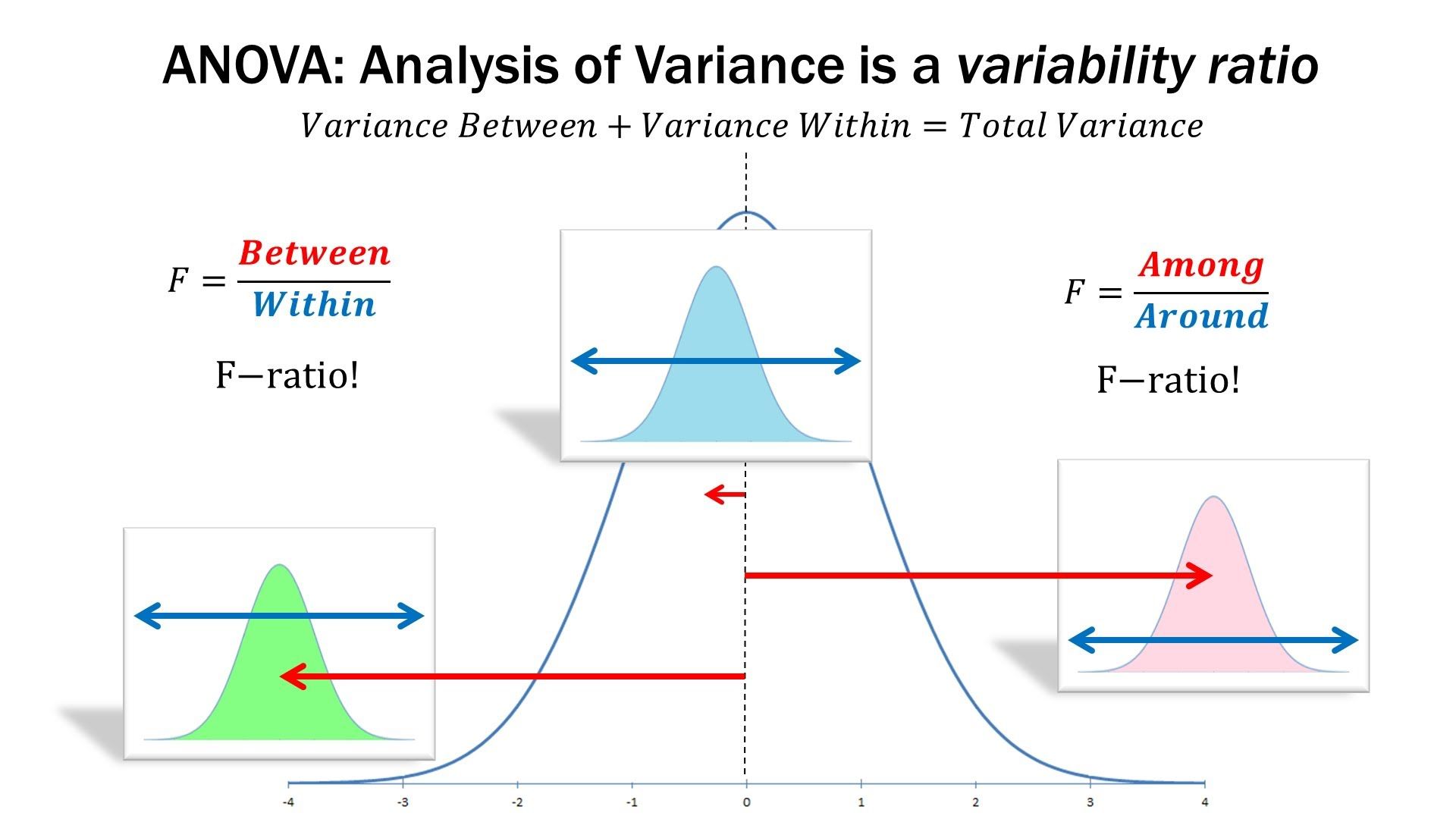
估计阅读时长: 14 分钟一般而言,如果我们在进行组学数据分析的时候,如果想要比较两组数据之间是否存在有差异性,一般是对两两比较的两组数据进行T-检验。但是在代谢组学数据分析领域内,则很多的组学数据分析情况为比较两组以上的数据,寻找差异的biomarker。那这个时候就需要使用上ANOVA统计检验方法了。 Order by Date Name Attachments anova • 105 kB • 874 click 2022年5月28日ANOVA-screen • 27 […]
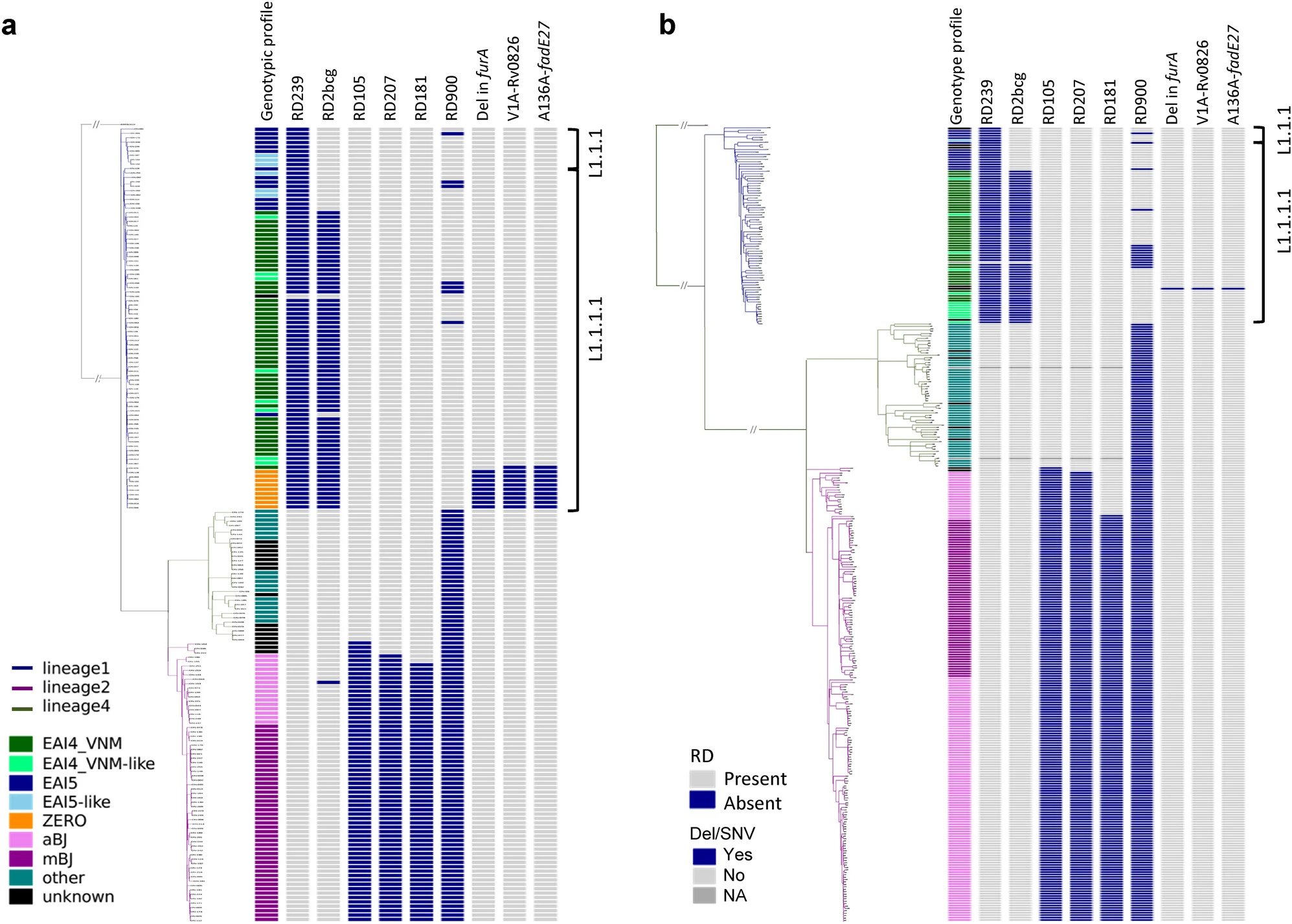
估计阅读时长: 7 分钟F统计量是群体遗传学中由Sewall Wright提出的重要统计量,用于衡量遗传变异在群体中的分布情况。它提供了对群体遗传结构和遗传分化的定量描述。F统计量主要有三种类型:Fis、Fit和Fst,分别反映个体内的、总体的和群体间的遗传分化。F统计量在群体遗传学中通常指的是Fst(Fixation Index,固定指数),它是一个衡量群体间遗传差异的指标。Fst的值范围从0到1,其中0表示群体间没有差异,1表示群体间完全分离。在群体遗传学研究中,Fst常用于评估群体的遗传多样性、群体间的迁移率以及自然选择的压力等。 Order by Date Name Attachments 41598_2021_92984_Fig1_HTML • 2 MB • 746 click 2022年5月28日p1 […]
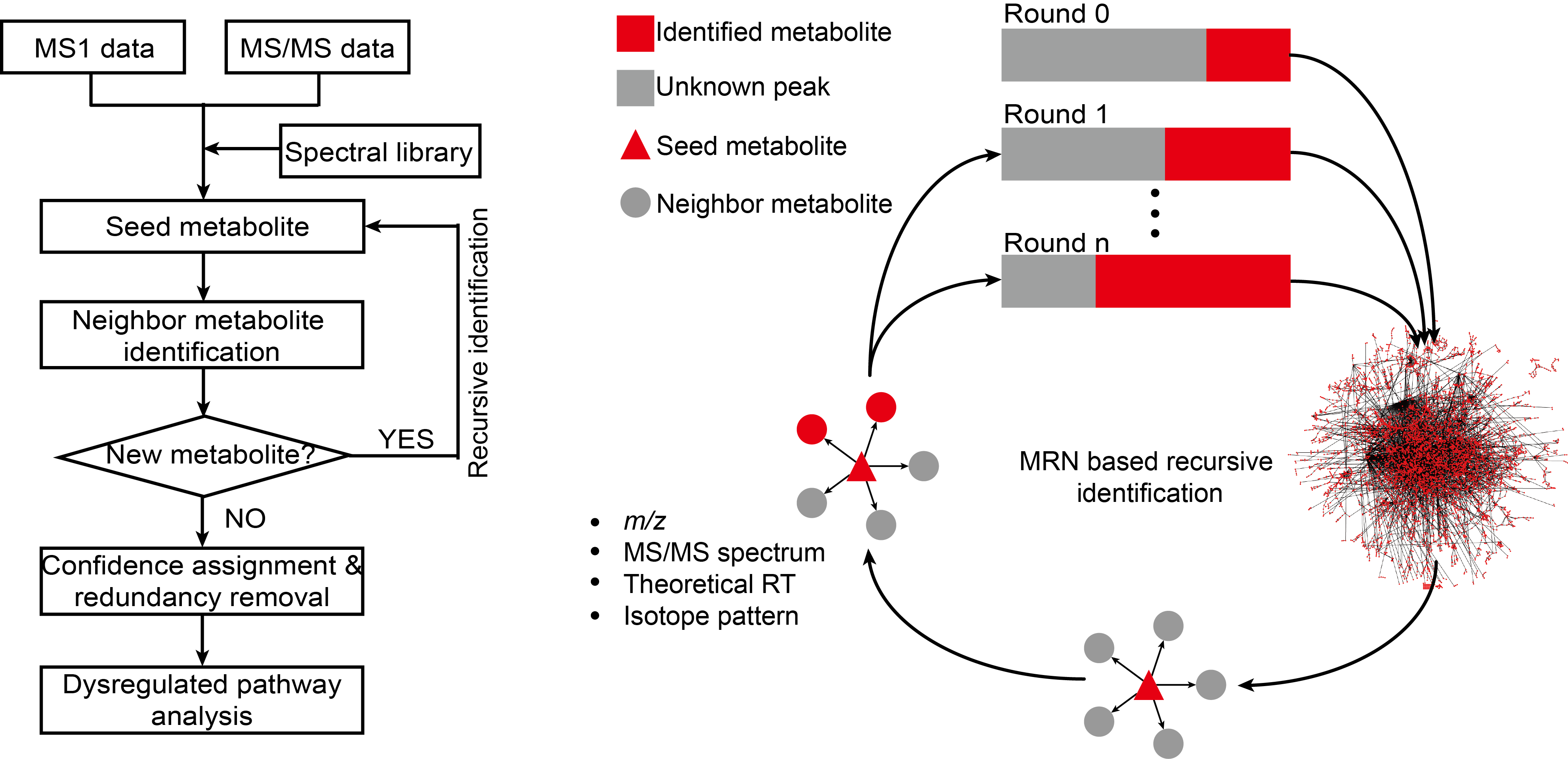
估计阅读时长: 6 分钟访问在线服务: http://metdna.zhulab.cn/ Metabolite identification is the long-standing challenge for liquid chromatography-mass spectrometry (LC-MS)-based untargeted metabolomics. Here, […]
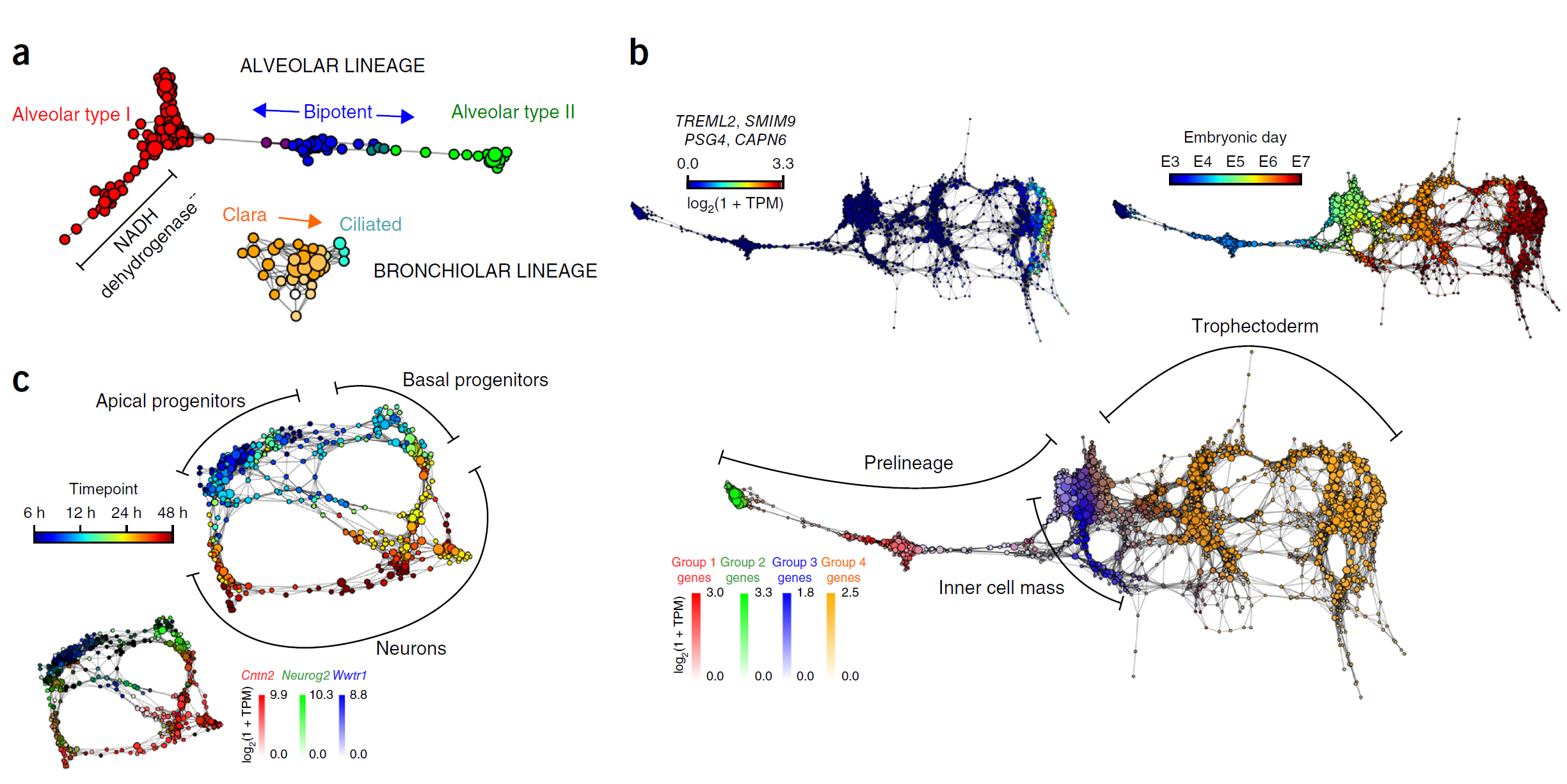
估计阅读时长: 14 分钟单细胞分析方法学习文献打卡记录: 【单细胞组学】PhenoGraph单细胞分型 【单细胞分析方法】VeTra:基于RNA速度的轨迹推断工具 【单细胞分析方法】单细胞图嵌入 Order by Date Name Attachments Cellular populations during motor neuron differentiation • […]
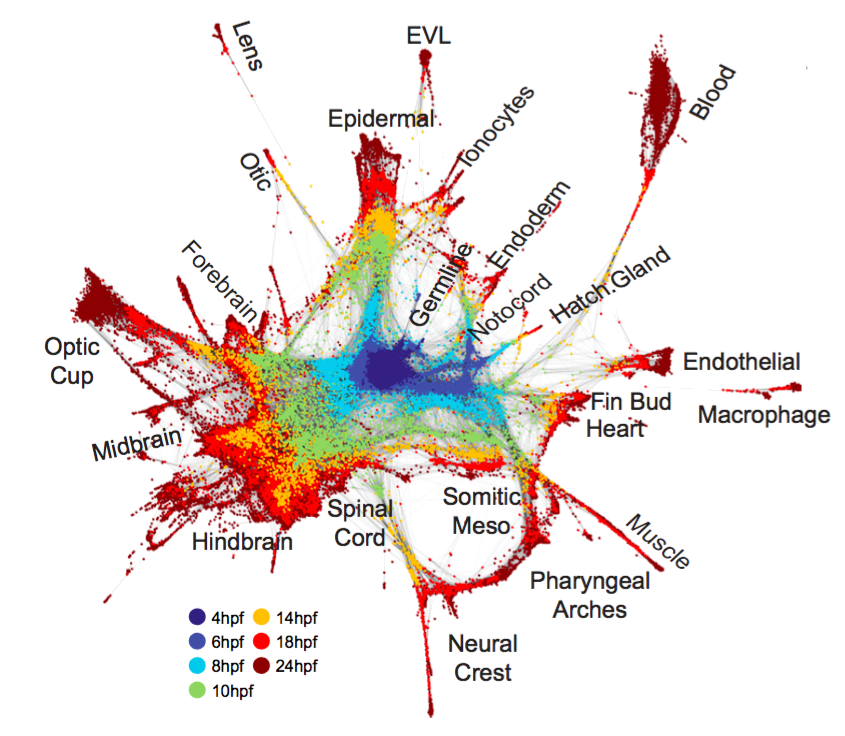
估计阅读时长: 7 分钟Assembles a manifold that is defined through a series of overlapping, locally-defined PCA subspaces. Non-mutual k-nearest-neighborhoods […]

估计阅读时长: 5 分钟https://github.com/xieguigang/graphQL 构建一个图数据库,可以用来帮我们解决复杂的知识关联计算问题。例如我们想要程序向我们回答dihydrogen oxide与water是否是同一个东西。如果光从字符串比较角度上面来看待这个问题的话,很显然,二者的字符串比较结果肯定是False。面对上面的这个问题,图数据库则可以很简单的向我们回答道上面的两个字符串都是指代的同一个东西。 Order by Date Name Attachments tumblr_inline_mqvdlydGCp1qz4rgp • 124 kB • 644 click 2022年3月5日Capture […]
估计阅读时长: 15 分钟https://gcmodeller.org 在这篇博客文章之中,我主要是来详细介绍一下是如何从头开始实现Phenograph单细胞分型算法的。在之前的一篇博客文章《【单细胞组学】PhenoGraph单细胞分型》之中,我们介绍了Phenograph算法的简单原理,以及一个在R语言之中所实现的Phenograph算法的程序包Rphenograph。在这里我主要是详细介绍在GCModeller软件之中所实现的VisualBasic语言版本的Phenograph单细胞分型算法。 Attachments Rphenograph • 236 kB • 694 click 2021年9月20日
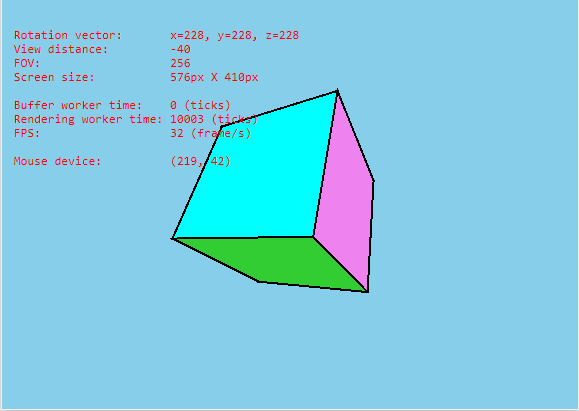
估计阅读时长: 17 分钟https://github.com/xieguigang/sciBASIC/tree/master/gr/Microsoft.VisualBasic.Imaging/Drawing3D 因为大家大多数都是从小接受电子游戏,所以长大了之后能够自己从零开始开发一个完整的3维图形引擎是每一个男程序员的梦想。这个就像玩机械的男人的梦想就是自己从头开始组装一辆汽车。还好这个梦想我在几年前就已经实现了。 Order by Date Name Attachments Cube3D_VB.NET • 4 MB • 776 click 2021年9月19日Cube_screenshot • […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?