
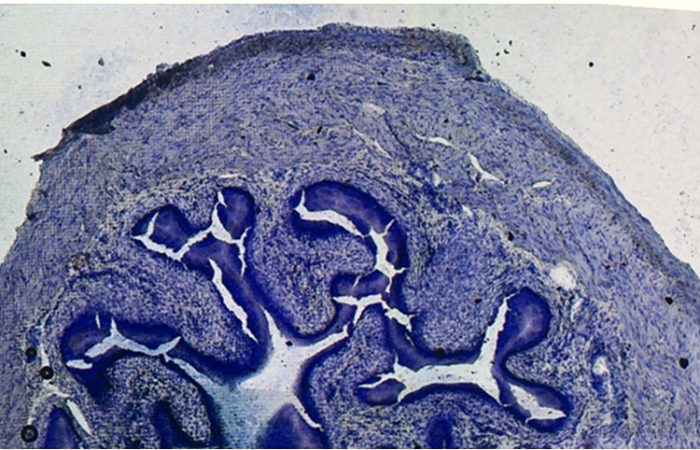
估计阅读时长: < 1 分钟HE染色(苏木精-伊红染色,Hematoxylin and Eosin Staining)是一种组织学和病理学中广泛应用的技术,用于染色组织切片以便于显微观察。该技术利用两种染料——苏木精和伊红,分别染色细胞核和细胞质,从而呈现出清晰的细胞结构。用于观察组织和细胞的形态结构。HE染色技术因其简单、高效的特点,成为组织学、胚胎学和病理学研究中不可或缺的基础方法。HE染色通过苏木精和伊红的染色作用,帮助观察和区分细胞核与细胞质及细胞外基质的形态结构。解读HE染色结果需要结合实验背景和研究目的,观察染色的均匀性、对比度以及细胞和组织的形态变化,从而提供重要的实验依据。 HE染色技术的历史可以追溯到19世纪末: 1868年:德国病理学家Karl Weigert开发了苏木精染色法,用于染色细胞核。 1877年:Albrecht von Leube开发了伊红染色法,用于染色细胞质。 1876年:化学家Wissowzky首次联合使用苏木精和伊红,但未被认为是发明者。 1886年:Paul Ehrlich发表了相关研究,推动了HE染色技术的标准化和广泛应用。
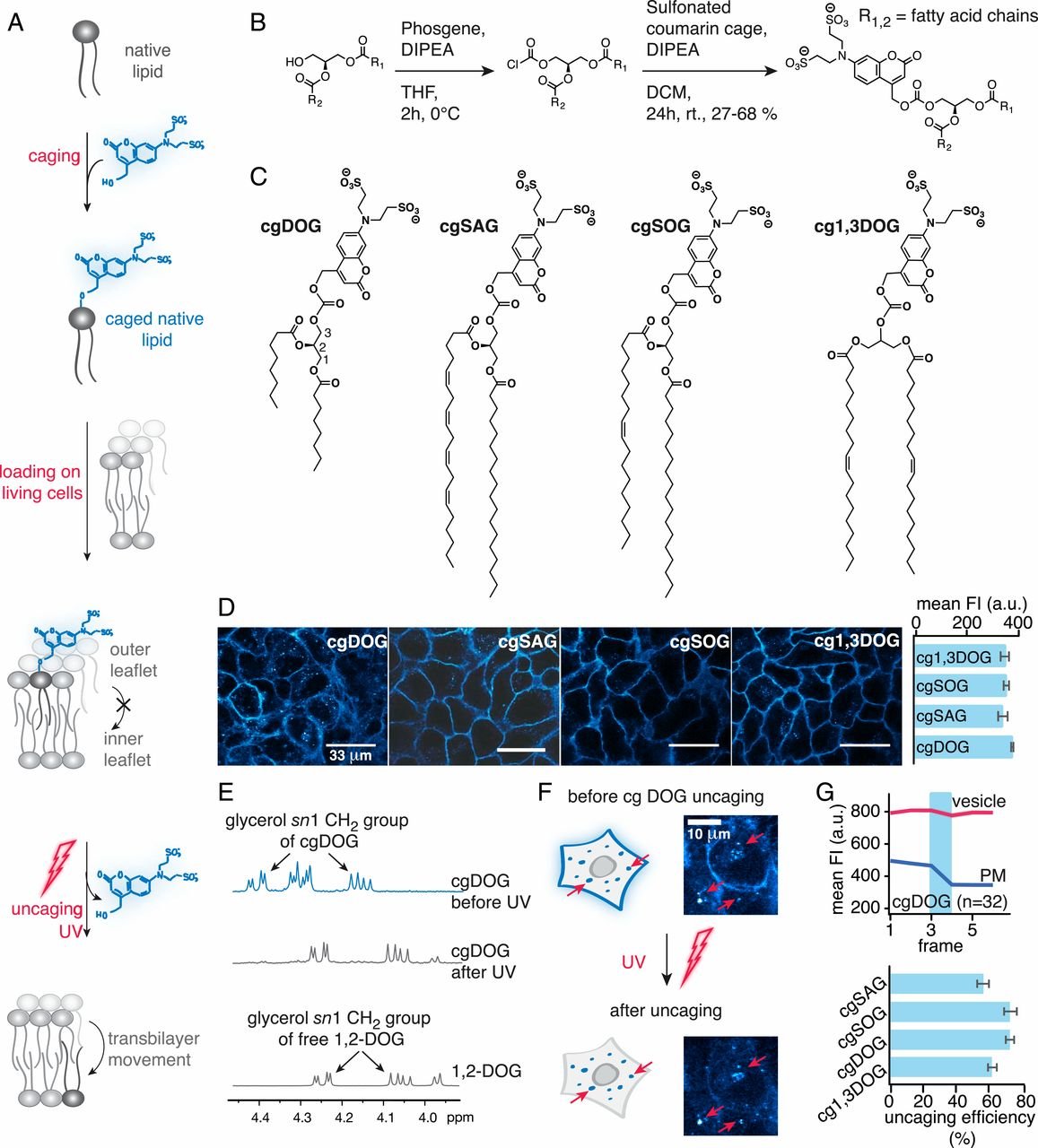
估计阅读时长: 2 分钟脂质组学作为系统生物学和代谢组学的重要分支,近年来取得了长足的发展,已成为生命科学研究中不可或缺的重要工具。 脂质组学的定义与研究内容 脂质组学是系统研究脂质组的一门独立学科,作为大规模定性和定量研究脂类化合物并了解它们在不同生理、病理条件下的功能和变化的方法学,能准确全面地提供生物样品中的脂质信息。它被定义为对生物体、组织或细胞中的脂质以及与其相互作用的分子进行全面系统的分析、鉴定,进而揭示脂质代谢与细胞、器官乃至机体生理病理过程的关系。 脂质是一类具有疏水性并且在大多数情况下可以溶于有机溶剂的物质,当然,还有部分脂质因为带有极性基团,往往是亲水性的,例如磷脂等。脂质组学是对生物体内的脂质进行系统分析的一门新兴学科,是代谢组学的重要分支。基于液质联用技术(LC-MS),无偏向性、尽可能多地检测细胞、组织、器官或体液等生物样本中的脂质。
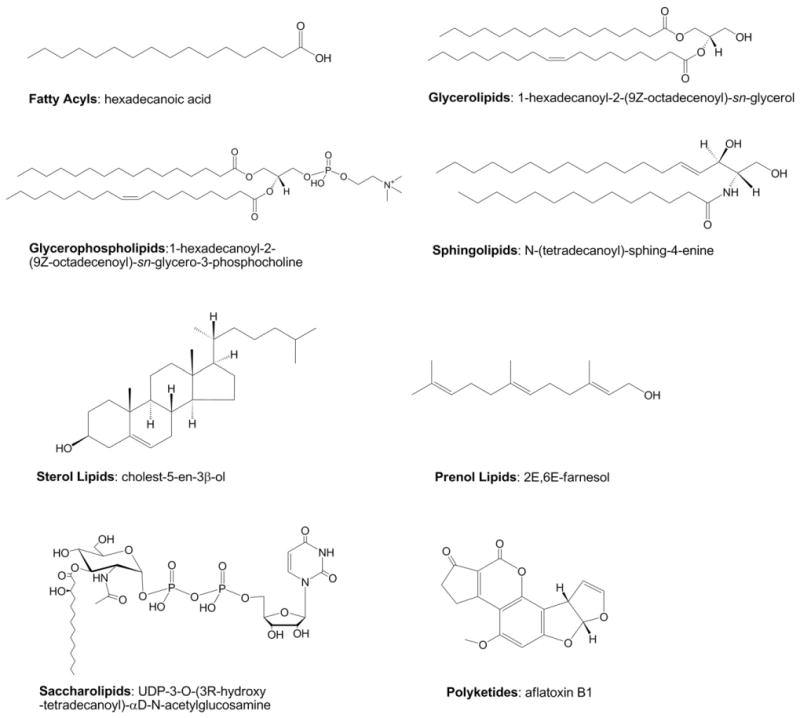
估计阅读时长: 3 分钟引言 二酰甘油(Diacylglycerol,DAG)是细胞内一类重要的脂质分子,作为生物膜的重要组成部分,同时也参与多种生物学功能和代谢途径。Diacylglycerol (DAG, 16:0_18:1) 是甘油二酯的一种异构体,其结构为sn-1位连接棕榈酸(16:0)、sn-2位连接油酸(18:1)。它在脂质代谢中具有多重角色,涉及合成、分解、信号传导及能量调节等过程。

估计阅读时长: 4 分钟假设我们现在拥有魔兽世界游戏的GM权限,那么我们可以怎样来组织一场有一千个玩家参加的PvP战斗呢? Order by Date Name Attachments 762191341 • 619 kB • 538 click 2025年6月7日AddonList • 35 […]

估计阅读时长: 8 分钟为了能够稳定的使用Ubuntu系统作为我的测试环境,我专门安装了Ubuntu的2404 LTS版本。但是发现虽然是LTS版本,仍然会因为系统更新而出现各种莫名奇妙的BUG。例如,在今天我为了安装软件,使用apt upgrade更新系统环境之后,出现了下面的消息: Order by Date Name Attachments ubuntu-24-04-noble-numbat • 66 kB • 487 click 2025年6月7日ti8S1 […]

估计阅读时长: 10 分钟 https://github.com/rsharp-lang/athena 在这博客篇文章中的Idea来自于最近进行AI Agent的开发:希望可以有一个方便的框架,能够让我像写普通的R#程序包的方式,为大语言模型编写对应的工具包,实现AI Agent功能。经过了大概一天的摸索,搞定了这样子的AI Agent的框架。我将这个框架取名为雅典娜系统。关于这个系统的名称,我们首先来看看DeepSeek是怎么说的: “我现在正在开发一个和大语言模型助手相关的工具,这个工具可以完成通过自然语言聊天的方式调用相应的工具完成生信数据分析,我需要为这个项目取一个名称,要求这个项目的名称应该是符合下面的需求的:女孩英文名称,和智慧相关的,和科学相关的。为我列举一些候选名称” 根据你的需求(女孩英文名、智慧与科学相关),结合搜索结果中的历史文化名人和寓意,以下是推荐的候选名称及来源解析: 名字 寓意 关联 Athena 源自希腊神话的智慧女神,象征智慧与科学战略。 雅典娜不仅是智慧化身,也代表理性思维,适合强调数据分析与策略的项目。 Clara 拉丁语“明亮、清晰”,象征清晰的科学思维。 […]
估计阅读时长: 8 分钟 https://github.com/xieguigang/LLMs 已知,现在我们可以成功的和正在运行的大语言模型服务勾搭了,现在能够让大语言模型为我们做些什么。很遗憾的是,由于大语言模型本质上只是一个数学模型,其作用只是针对我们的输入找出最佳的字符输出组合。如果我们没有额外的针对大语言模型进行拓展,我们所勾搭上的大语言模型充其量也只是一个聊天机器人,他既不能帮我们发送email,也不能够帮助我们调节屋内的灯光,只能够做到分析我们输入的文本,然后输出一段最佳的文本。所以我们需要通过针对大语言模型添加额外的拓展来帮助我们实现各种功能。 又已知,大语言模型的本质就是进行文本的结构化分析,那么假如我们的输入信息中包含有某些工具函数的描述信息,而且大语言模型能够正确的分析出我们的输入文本和输入信息中所包含的工具函数之间的对应关系,那么大语言模型的输出就可以专门定向的变换为一种针对输入信息所对应的函数调用的结构化文本信息输出。当运行大语言模型的基础服务捕捉到这种结构化文本(例如json)输出后就可以通过这种结构化文本信息的内容解析结果来调用对应的外部工具,这样子我们就可以让大语言模型来帮助我们完成特定的任务了。这种特性就是大语言模型的Function Calling功能。
估计阅读时长: 10 分钟 https://github.com/xieguigang/LLMs 大语言模型从2023年开始,在最近几年非常的火爆。在最近的一段时间,有大语言模型自动化处理数据的需求,开发了一个基于Ollama服务的客户端来通过大语言模型执行自动化任务。在这里记录下这个开发过程。 Ollama介绍 Ollama 是一个开源的大型语言模型(LLM)服务工具,专注于简化本地环境中大模型的部署与管理。它通过类似 Docker 的框架设计,让用户能以极低门槛在个人电脑或服务器上运行各类开源模型(如 Llama 3、Mistral、DeepSeek 等),实现数据隐私与离线推理的平衡。 Order by Date Name Attachments […]
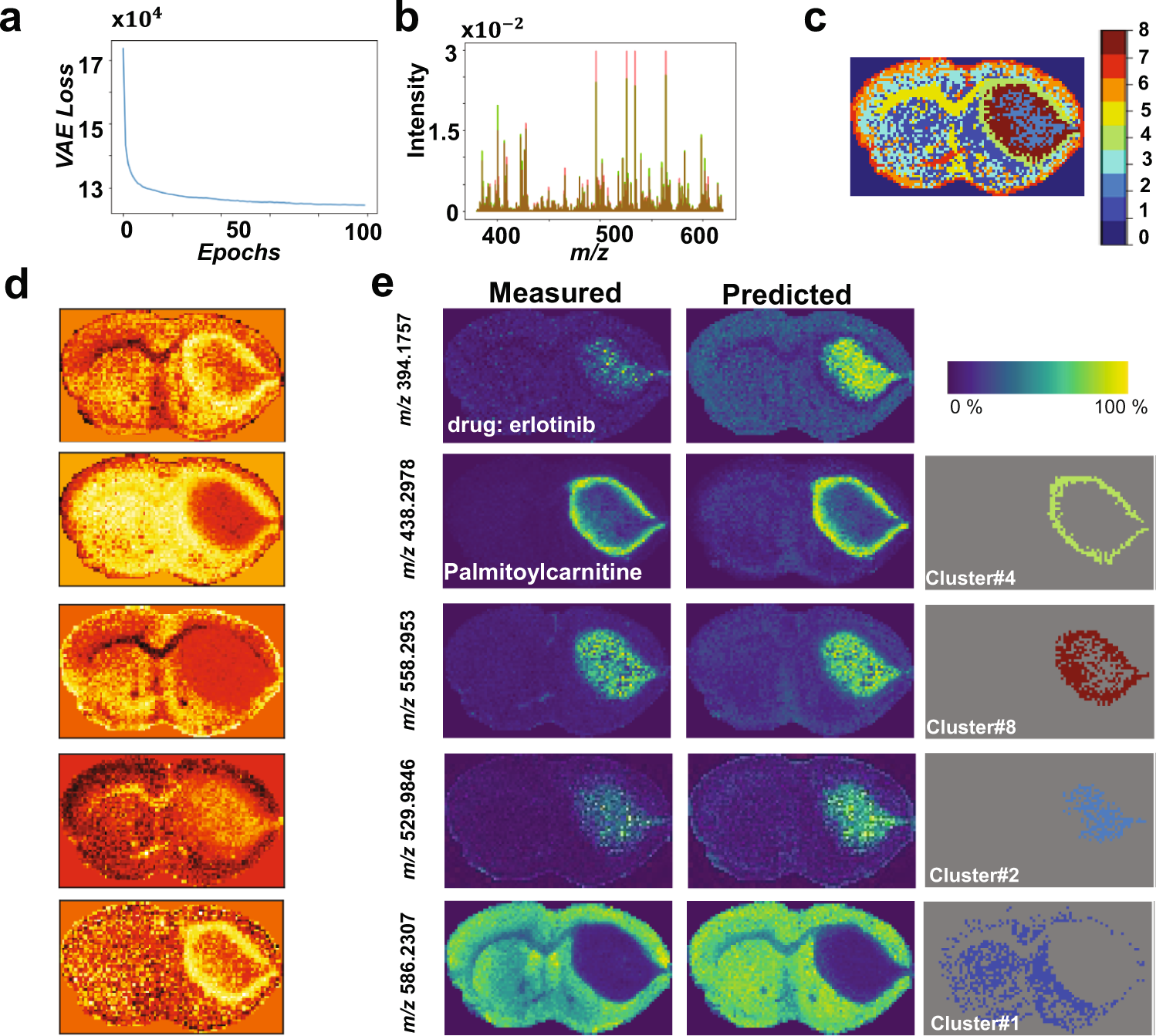
估计阅读时长: 4 分钟基于UMAP工具进行简单的自动化组织分区操作 在这里我们假设已经可以正常的将空间代谢数据导入至MZKit工作站软件之中。假若需要借助于MZKit工作站软件进行切片组织样本的自动化分区操作,相关的功能可以在【MSI Analysis】菜单栏中寻找到。在这里我们打开【Show Map Layer】按钮,选择【UMAP and clustering】功能。 基于降维的组织自动化分区原理 因为降维操作一般是一种特征提取操作,所以经过降维之后,在高维度空间上无法显现的特征,在低维度会呈现出来。在高维度空间散落的相近的数据点,在经过特征提取之后,低维度上会产生相似的特征信息,相互聚集在一簇。这样子我们就可以在低维度空间上通过一些聚类算法讲这些特征进行聚类,最后将聚类特征结果标记到各个散点上的对应的原始成像空间上,我们就可以看见组织分区的结果了。 Abdelmoula, W.M., Lopez, B.GC., Randall, E.C. et […]
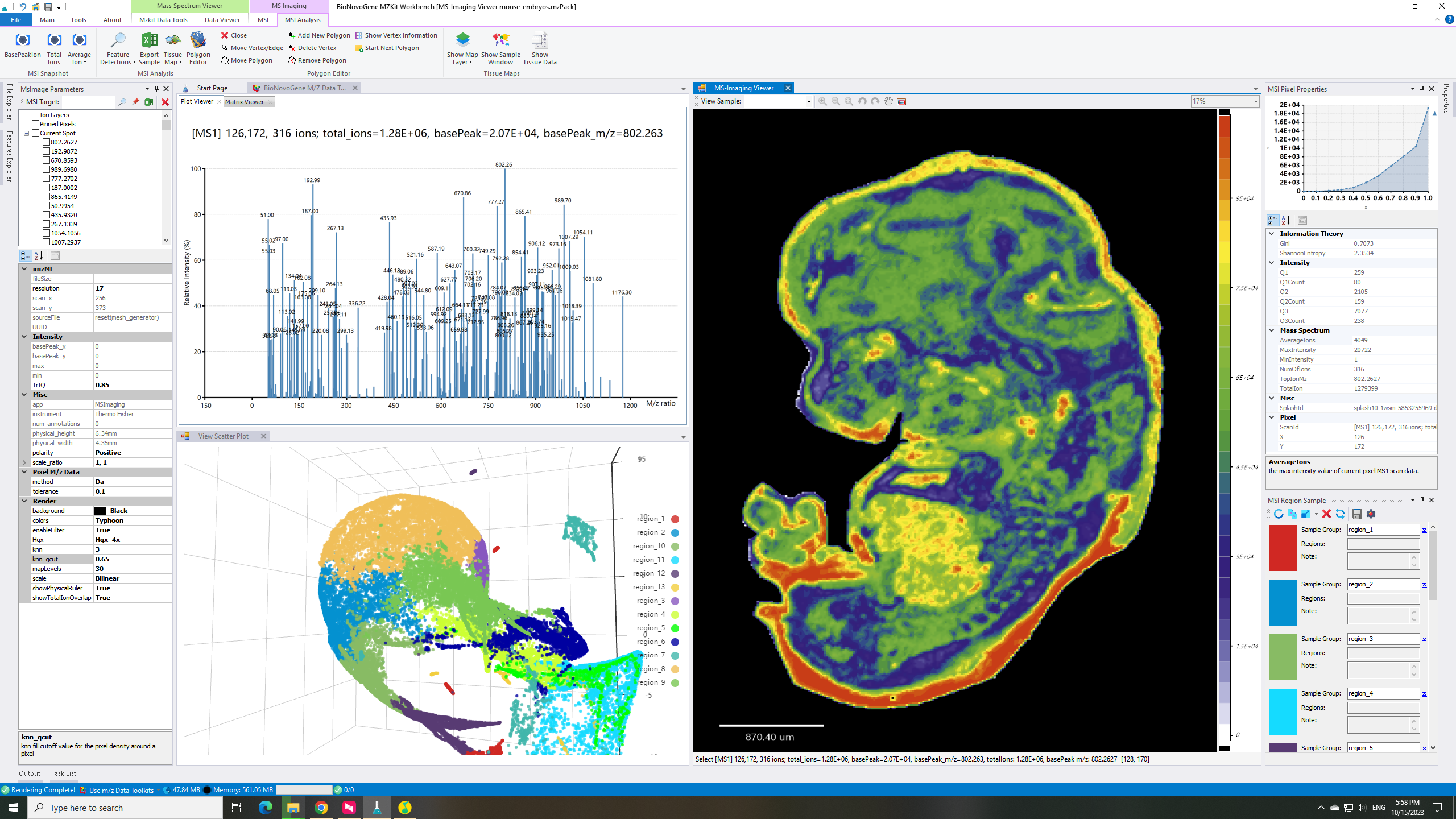
估计阅读时长: 6 分钟大家好呀,今天的这篇文章主要是为了回答在B站上的一位小伙伴的请求 Order by Date Name Attachments render-parameters • 18 kB • 640 click 2023年10月15日view-umap • 427 […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?